








2025年无菌制药法规变革深度解析
摘要: 近日,国家药品监督管理局(NMPA)综合司发布《药品生产质量管理规范》无菌药品附录(征求意见稿),标志着我国无菌制药监管即将迎来史上最严变革。此次修订被业内称为‘中小型企业生死劫’,不仅在结构上进行了系统 ...
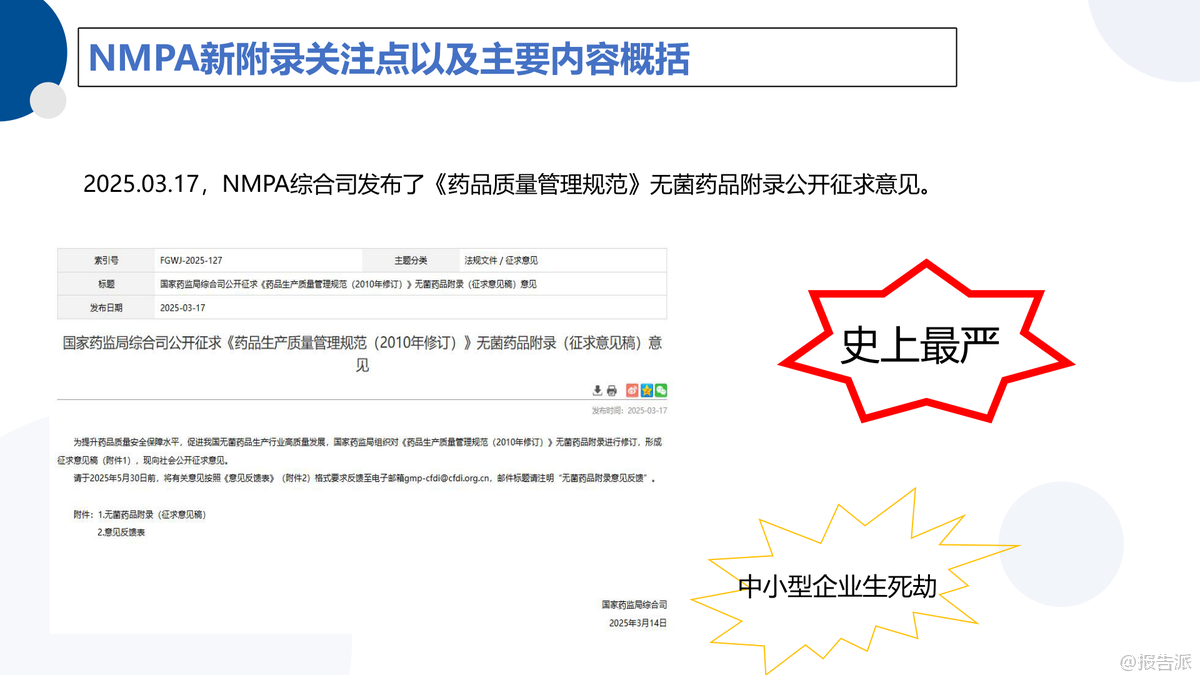
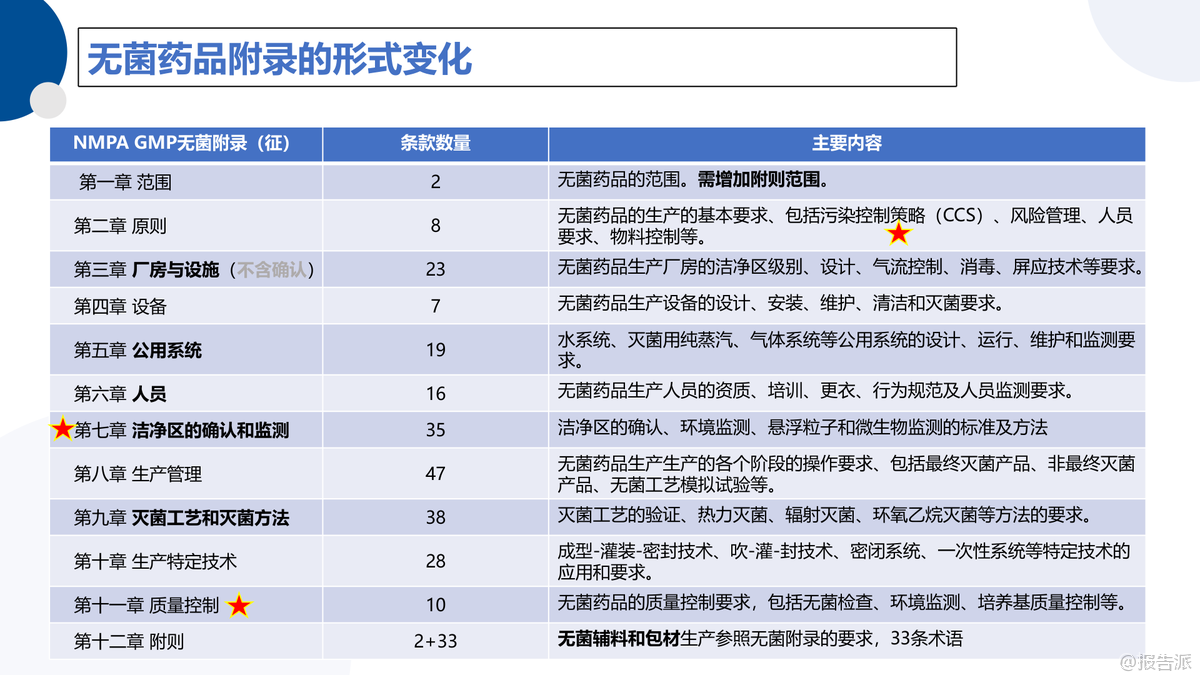
| 近日,国家药品监督管理局(NMPA)综合司发布《药品生产质量管理规范》无菌药品附录(征求意见稿),标志着我国无菌制药监管即将迎来史上最严变革。 此次修订被业内称为‘中小型企业生死劫’,不仅在结构上进行了系统性重构,更在理念上实现了从‘合规驱动’向‘风险防控与工艺本质安全’的深刻转变。 新版附录共15章、80条条款,相较2010版的12章、235条大幅精简,但内容更为聚焦与深化。 其核心变化在于确立了“无菌保障”和“污染控制策略(CCS)”为贯穿药品全生命周期的核心主线。 这意味着企业不能再依赖末端检测或人为补救,而必须从前端设计、硬件配置、工艺流程和人员行为等多维度构建系统性防护体系。 值得关注的是,新附录明确提出优先通过硬件设施保障无菌环境,而非过度依赖制度流程和人员自觉。 这预示着未来无菌制药将加速向自动化、密闭化、隔离化方向发展,RABS(限制进出屏障系统)、隔离器、一次性使用系统(SUS)等先进技术的应用将成为标配。 同时,对A/B级洁净区的风速、气流流型、自净时间等关键参数提出了更严格的验证要求,并强调气流可视化研究需覆盖动态模拟操作场景,确保实际生产中的污染风险可控。 在验证维度上,新版附录强化了洁净区确认与环境监测的区分。 洁净区级别确认需基于ISO 14644标准,涵盖静态与动态两种状态下的悬浮粒子和微生物测试,且A/B级区域再确认周期不得超过6个月。 特别指出的是,D级区动态粒子限度不再统一规定,企业需基于风险评估自行制定标准,体现了监管的科学性与灵活性。 此外,新附录首次将密闭系统、一次性系统(SUS)、吹灌封(CFS)等新兴技术纳入正式条款,并明确PUPSIT(培养基模拟灌装试验)的技术要求。 对于水系统、除菌过滤、灭菌工艺等关键环节,也进一步细化了完整性测试、使用后检测、冗余过滤判定等操作规范,尤其强调除菌过滤单元的完整性测试必须在使用后立即执行。 总体来看,2025版无菌药品附录不仅是技术标准的升级,更是监管理念的跃迁。 它推动企业从被动应对转向主动预防,从经验管理迈向数据驱动。 对于制药企业而言,这既是挑战也是机遇——唯有尽早布局CCS体系建设,强化硬件投入与验证能力,才能在这场行业洗牌中立于不败之地。 出品方:中电二 发布时间:2025年
|
推荐文章

2
2025年云计算行业应用场景报告
资讯
76人已阅读
3
2025年文科生AI编程研究报告
资讯
78人已阅读

4
2025年人工智能与进攻性安全研究报告
资讯
81人已阅读

5
2025年数据库行业技术趋势报告
资讯
77人已阅读

6
2025年生成式人工智能商业价值报告
资讯
73人已阅读

7
2025年体育领域政策汇编报告
资讯
73人已阅读
8
2025年大型央国企“十五五”战略规划编制实
资讯
95人已阅读

9
2025年电子元件供应链研究报告
资讯
92人已阅读

10
2024年Web3及金融科技研究报告
资讯
68人已阅读
数据图表

2
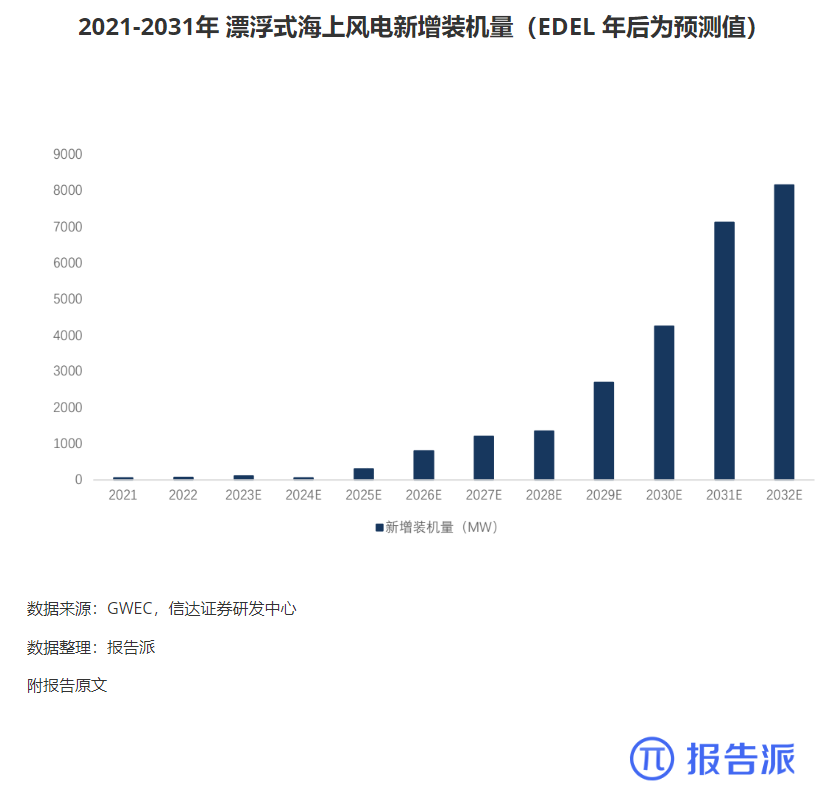
2011-2031 年全球海上风电装机量(含预测)
行业数据
1716人已阅读
3
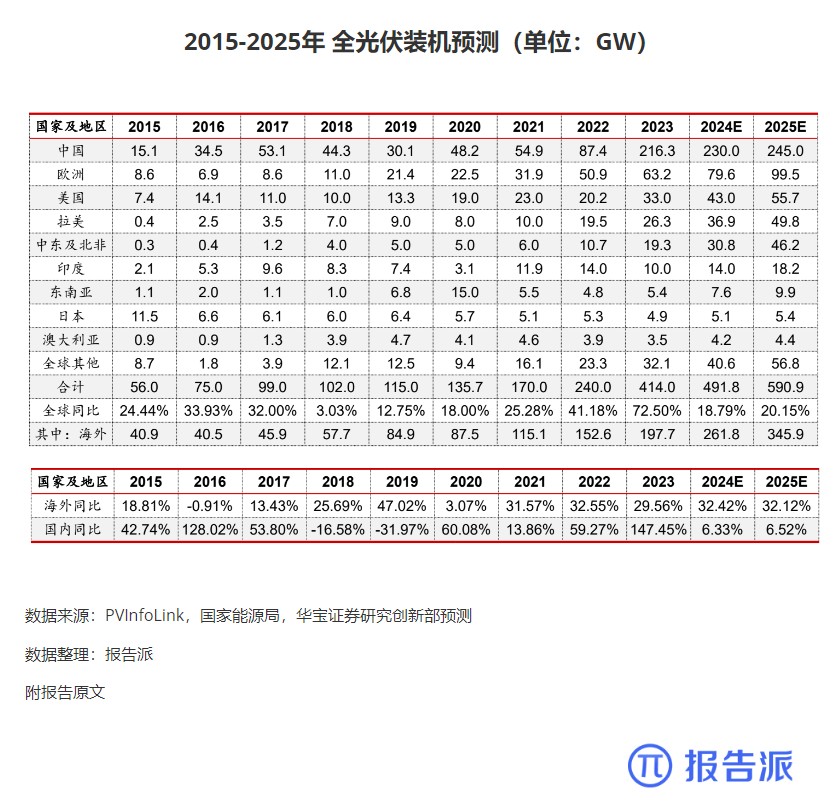
2015-2025年 全光伏装机预测(单位:GW)
市场规模
1949人已阅读
4
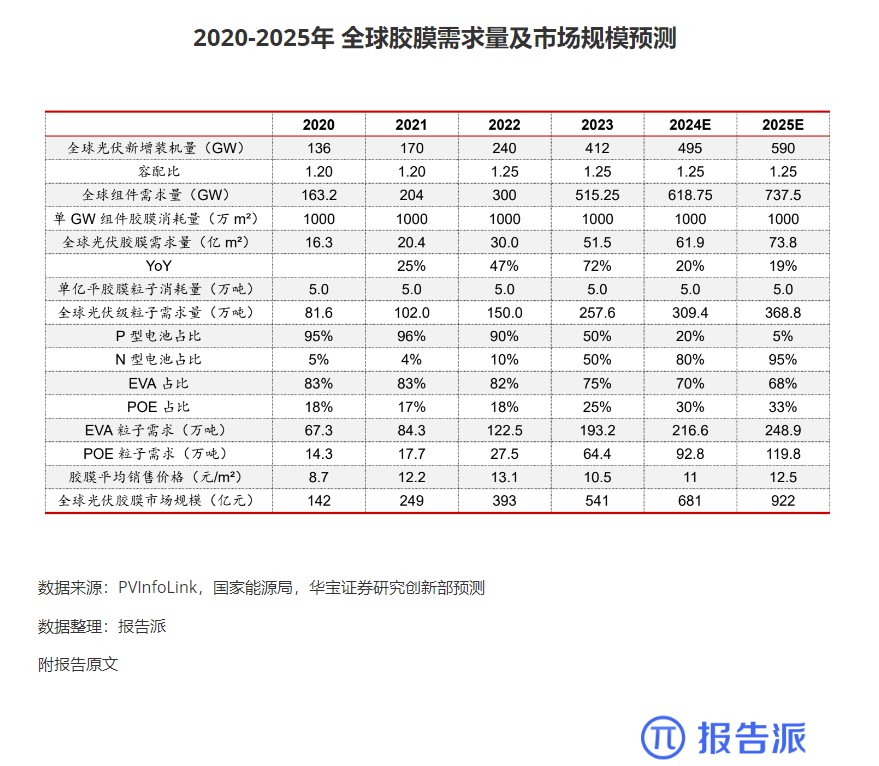
2020-2025年 全球胶膜需求量及市场规模预测
市场规模
1862人已阅读
5
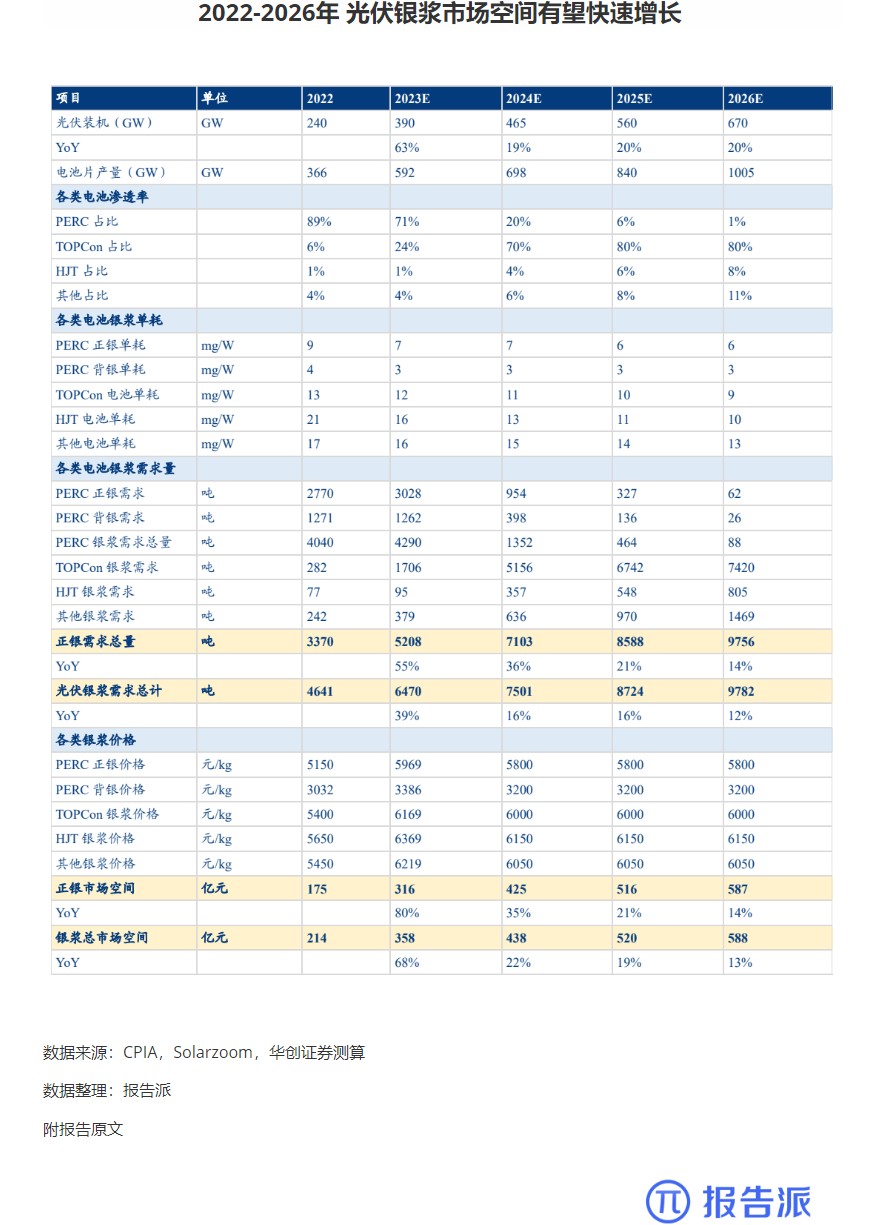
2022-2026年 光伏银浆市场空间有望快速增长
市场规模
1932人已阅读
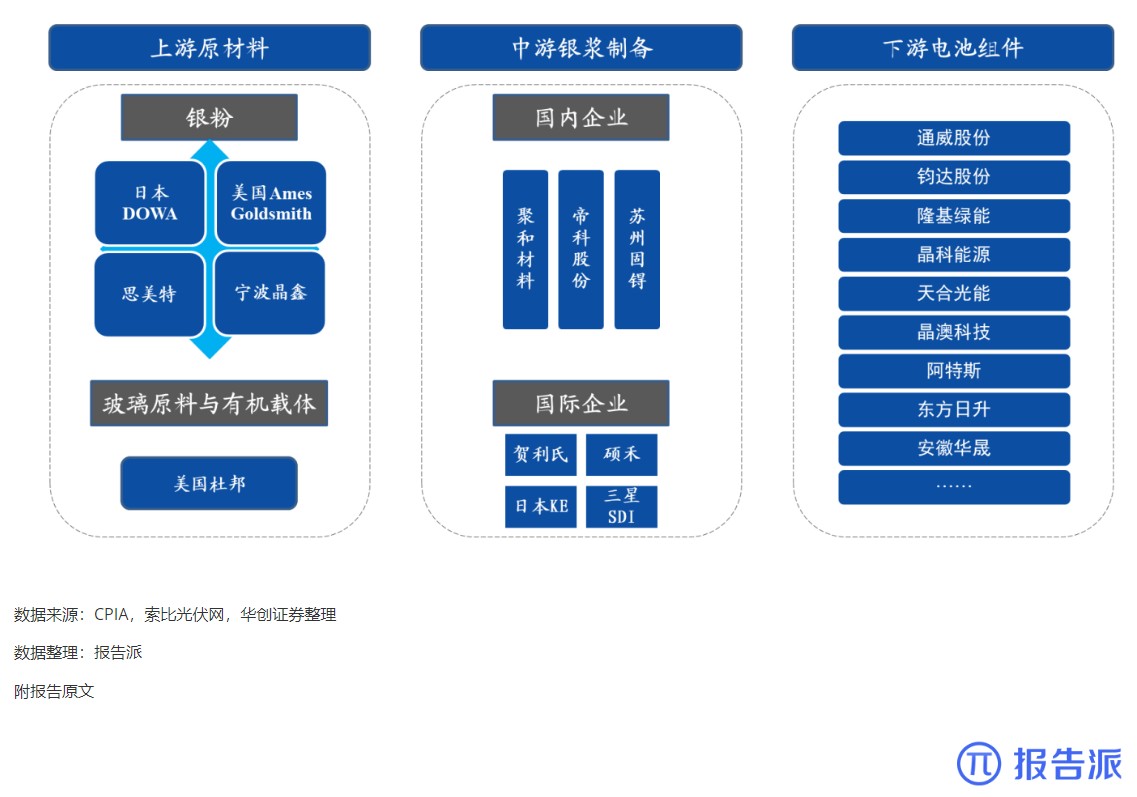
6
光伏银浆产业链相对简单
技术工艺
1828人已阅读
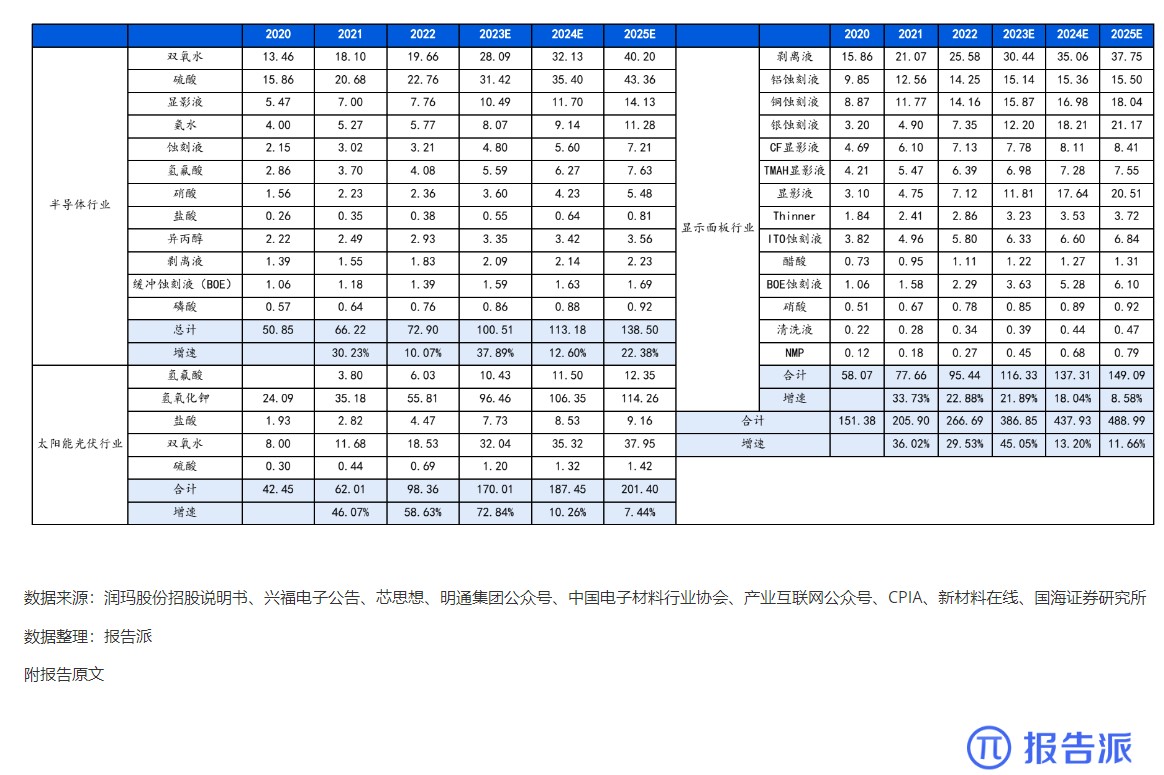
7
2020-2025年 我国湿电子化学品需求预测(万
市场规模
1813人已阅读
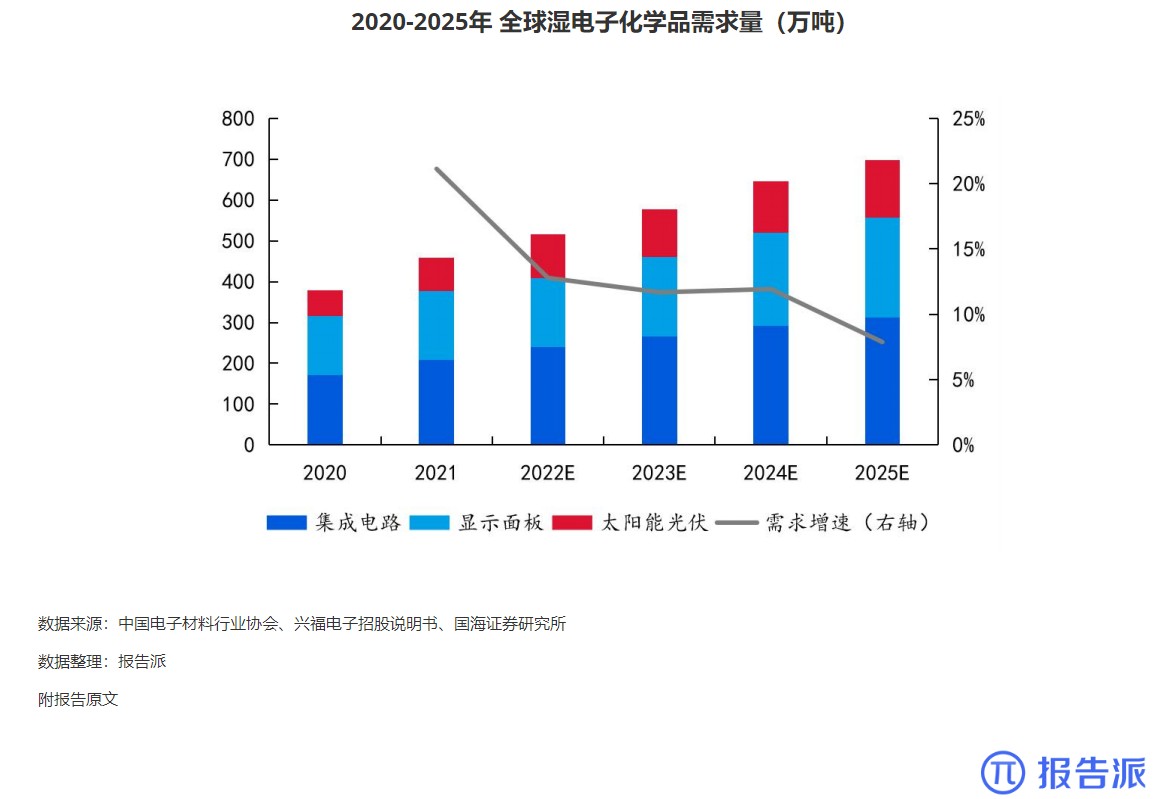
8
2020-2025年 全球湿电子化学品需求量(万吨
市场规模
1935人已阅读
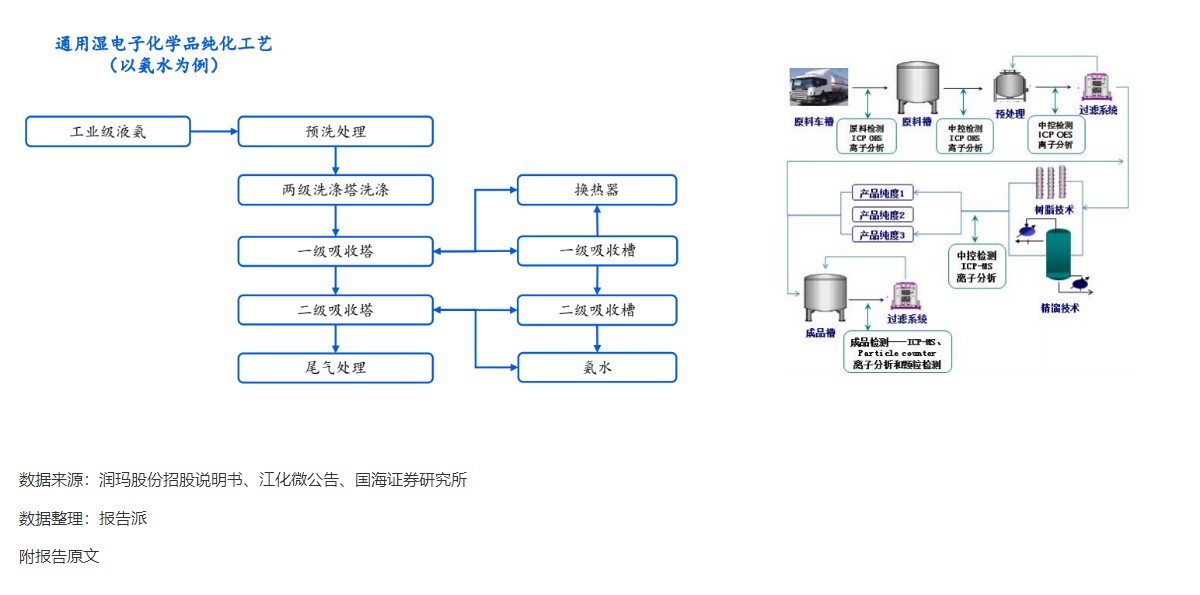
9
通用湿电子化学品纯化工艺
技术工艺
1673人已阅读
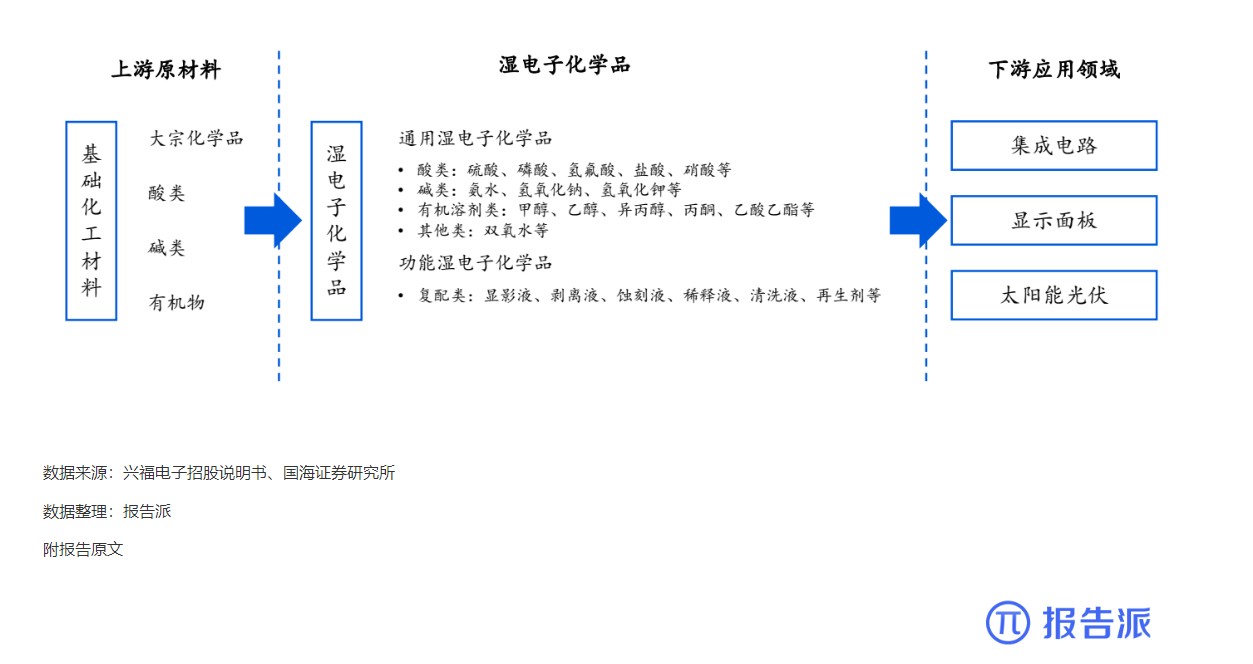
10
湿电子化学品上下游产业链基本情况
技术工艺
1944人已阅读
热门数据
1
2024年1—2月份规模以上工业增加值增长7.0%
2024-03-22
2
截至2023年底我国累计建成充电基础设施859.
2024-03-22
3
2024年3月21日人民币 NDF 远期合约汇兑美元
2024-03-21
4
2024年1—2月份能源生产情况
2024-03-21
5
2024年2月银行结售汇和银行代客涉外收付款
2024-03-21
6
2024年3月韩国方便面出口2.3万吨,同比增加
2024-03-21



